Accelerate Your Biomolecule
and Bioassay Research
and Bioassay Research
sales@neoscientific.com
+1-888.733.6849
+1-617.299.7367 (Int’l)
+1-617.299.7367 (Int’l)
+1-888.733.6849
+1-617.299.7367 (Int’l)
+1-617.299.7367 (Int’l)
For quotations, please use our online quotation form, and you may also contact us by
sales@neoscientific.com
+1-888.733.6849
+1-617.299.7367 (Int’l)
+1-888.733.6849
+1-617.299.7367 (Int’l)
| Reactivity | Human Mouse Rat |
| Tested applications | WB IHC IF |
| Recommended Dilution | WB 1:500 - 1:2000 IHC 1:50 - 1:200 IF 1:10 - 1:100 |
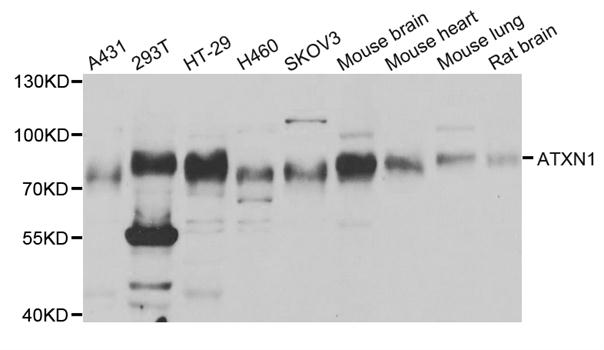
| Calculated MW | 87kDa |
| Observed MW | Refer to Figures |
| Immunogen | Recombinant protein of human ATXN1 |
| Storage Buffer | Store at -20℃. Avoid freeze / thaw cycles. Buffer: PBS with 0.02% sodium azide, 50% glycerol, pH7.3. |
| Synonym | ATX1; SCA1; D6S504E |
The autosomal dominant cerebellar ataxias (ADCA) are a heterogeneous group of neurodegenerative disorders characterized by progressive degeneration of the cerebellum, brain stem and spinal cord. Clinically, ADCA has been divided into three groups: ADCA types I-III. ADCAI is genetically heterogeneous, with five genetic loci, designated spinocerebellar ataxia (SCA) 1, 2, 3, 4 and 6, being assigned to five different chromosomes. ADCAII, which always presents with retinal degeneration (SCA7), and ADCAIII often referred to as the `pure' cerebellar syndrome (SCA5), are most likely homogeneous disorders. Several SCA genes have been cloned and shown to contain CAG repeats in their coding regions. ADCA is caused by the expansion of the CAG repeats, producing an elongated polyglutamine tract in the corresponding protein. The expanded repeats are variable in size and unstable, usually increasing in size when transmitted to successive generations. The function of the ataxins is not known. This locus has been mapped to chromosome 6, and it has been determined that the diseased allele contains 41-81 CAG repeats, compared to 6-39 in the normal allele, and is associated with spinocerebellar ataxia type 1 (SCA1). At least two transcript variants encoding the same protein have been found for this gene.
N/A



Copyrights © 2014 NEO Group 245 First Street, 18th Floor, Cambridge MA 02142 888.733.6849