Accelerate Your Biomolecule
and Bioassay Research
and Bioassay Research
sales@neoscientific.com
+1-888.733.6849
+1-617.299.7367 (Int’l)
+1-617.299.7367 (Int’l)
+1-888.733.6849
+1-617.299.7367 (Int’l)
+1-617.299.7367 (Int’l)
For quotations, please use our online quotation form, and you may also contact us by
sales@neoscientific.com
+1-888.733.6849
+1-617.299.7367 (Int’l)
+1-888.733.6849
+1-617.299.7367 (Int’l)
| Reactivity | Human Mouse Rat |
| Tested applications | WB IHC IF |
| Recommended Dilution | WB 1:500 - 1:2000 IHC 1:50 - 1:200 IF 1:50 - 1:200 |
| Calculated MW | 58kDa |
| Observed MW | Refer to Figures |
| Immunogen | Recombinant protein of human PSAP |
| Storage Buffer | Store at -20℃. Avoid freeze / thaw cycles. Buffer: PBS with 0.02% sodium azide, 50% glycerol, pH7.3. |
| Synonym | FLJ00245; GLBA; MGC110993; SAP1; |

Western blot analysis of extracts of various cell lines, using PSAP antibody.

Immunohistochemistry of paraffin-embedded human liver using PSAP antibody at dilution of 1:200 (x400 lens)
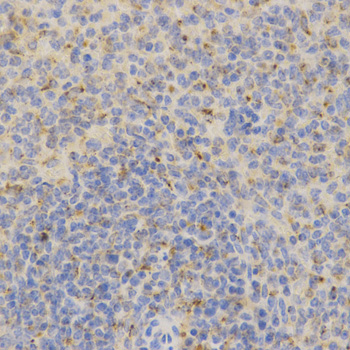
Immunohistochemistry of paraffin-embedded mouse spleen using PSAP antibody at dilution of 1:200 (x400 lens)
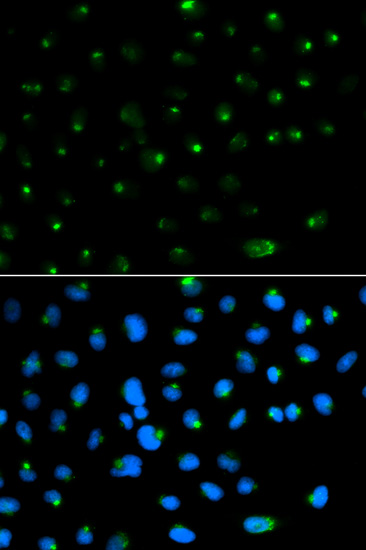
Immunofluorescence analysis of MCF7 cell using PSAP antibody. Blue: DAPI for nuclear staining.
The PSAP gene encodes prosaposin, a precursor of four small nonenzymatic glycoproteins termed 'sphingolipid activator proteins' (SAPs) that assist in the lysosomal hydrolysis of sphingolipids. After proteolytic processing of the presaposin protein, these 4 released polypeptides are functional activators. Saposin A is encoded by residues 60 to 143 of PSAP, saposin B by 195 to 275, saposin C by 311 to 390, and saposin D by 405 to 487. They are four 12-14 kDa heatstable glycoproteins. Saposins A-D localize primarily to the lysosomal compartment where they facilitate the catabolism of glycosphingolipids with short oligosaccharide groups. Saposins A-D are required for the hydrolysis of certain sphingolipids by specific lysosomal hydrolases. (PMID: 2001789) Defects in PSAP are the cause of Gaucher disease, Tay-Sachs disease, and metachromatic leukodystrophy (PubMed: 2060627, PMID: 15773042). This PSAP antibody (10801-1-AP) is expected to recognize both saposin A and B.
N/A



Copyrights © 2014 NEO Group 245 First Street, 18th Floor, Cambridge MA 02142 888.733.6849





